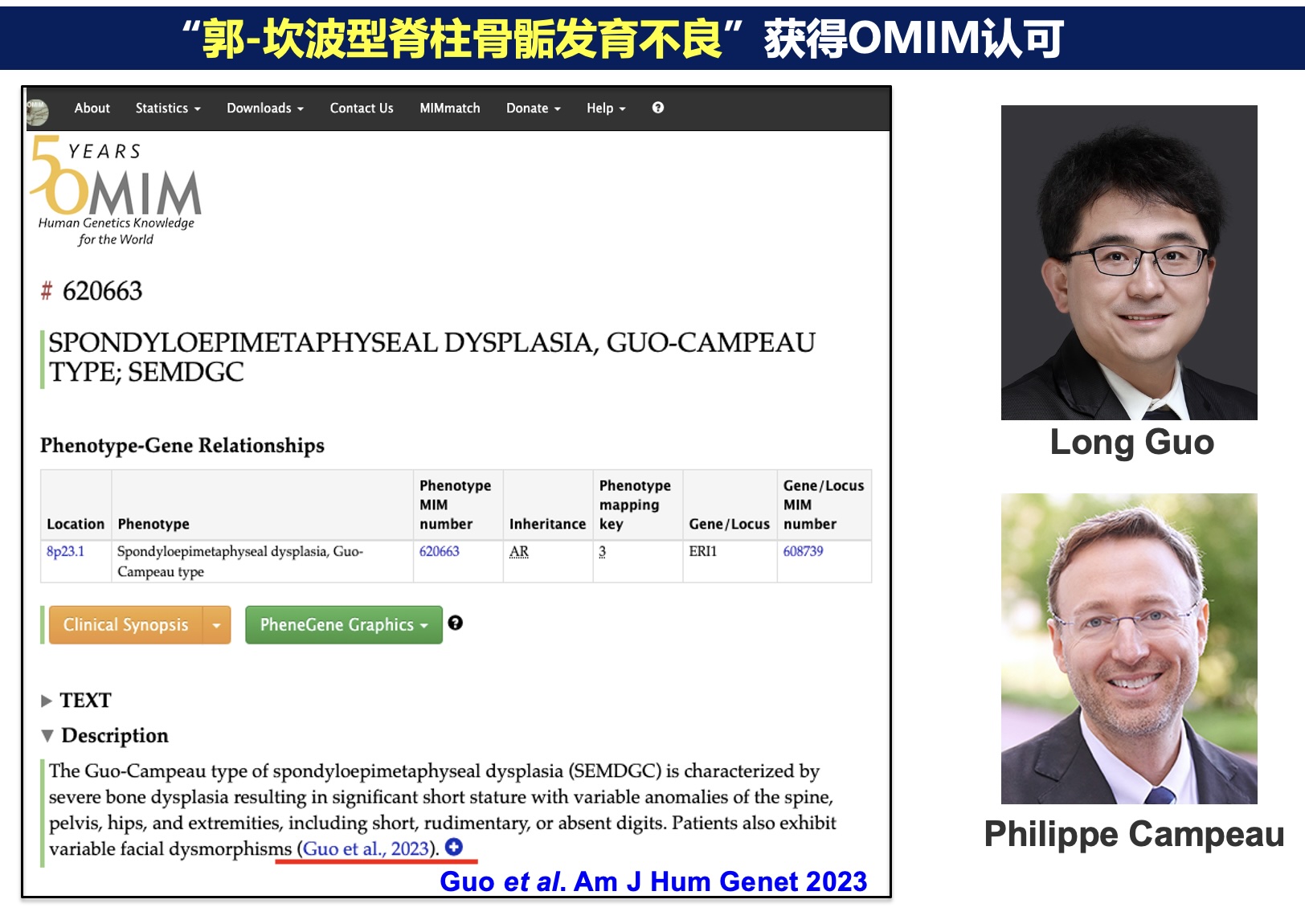
2023年6月22日,郭龙教授与加拿大蒙特利尔大学Philippe M Campeau教授合作,领衔国际罕见病多中心研究团队(由中国、加拿大、日本、德国、美国、英国、法国、南非的39家机构组成),在罕见病领域TOP杂志American Journal of Human Genetics上发表题为“Null and missense mutations of ERI1 cause a recessive phenotypic dichotomy in humans”(全文链接: https://authors.elsevier.com/c/1hIEegeX6Kob)的文章。该研究报道了郭龙教授团队发现的第5个新的人类疾病,将其命名为郭-坎波型脊柱骨骺发育不良(spondylo-epi-metaphyseal dysplasia(SEMD), Guo-Campeau type,OMIM #620663)(图1)。此外,该研究还first time提出一种隐性遗传病的表型二分类模型,为单基因遗传病“基因型与表现型关系”这一共性问题的探讨提供了新思路。 
图1 郭-坎波型脊椎骨骺发育不良的骨骼表型(从左到右可见: 扁平椎、长管状骨的骨骺及干骺端发育不良、指/趾畸形) 郭龙等在7个不相关家系中发现8位拥有类似表型的罕见病患者。通过外显子或基因组测序,发现患者均为ERI1基因的双等位变异。这些患者中,拥有至少一个错义变异的患者表现出以“SEMD+指/趾畸形”为特征的严重骨骼发育异常。对比之下,仅含有无功能变异的患者几乎没有指/趾以外的骨骼表型。这种错义变异较无功能变异引起更为严重表型的现象在显性遗传病中较为常见,但在隐性遗传病中尚属first time报道。ERI1编码一种核糖核酸外切酶,调控小鼠多种RNA的代谢,其在人体中的作用尚不清楚。为了阐明ERI1变异引起的生物学效应,郭龙等利用患者来源的诱导多能干细胞(iPSC)等工具进行了一系列功能学研究,发现患者iPSC形成软骨类器官的能力下降(图2),伴随5.8S核糖体RNA的成熟障碍以及细胞复制相关组蛋白信使RNA的降解缺陷(图3)。这些发现首次证实核糖核酸外切酶介导的RNA代谢过程对于人类软骨的发育和稳态维持至关重要,为软骨相关疾病治疗方法的开发提供了新线索。 
图2 患者由来iPSC向软骨类器官的分化减弱 
图3 郭-坎波型脊柱骨骺发育不良及其表型二分类模型的病理机制假说
该研究被以下媒体报道 https://medical.sciencenet.cn/sbhtmlnews/2023/7/368904.shtm?id=368904 http://www.med.xjtu.edu.cn/info/1246/14973.htm https://www.oldnormal.com/articles/3407;jsessionid=0A8900B358A4A8A2505D3FD939B08CFC https://min.news/health/a9cdefdc9e8d97835129add424a6f4bf.html http://m.gxcbt.com/lvyou/2023/0705/108131.html
|